Фенілкетонурія
Визначення - Що таке фенілкетонурія?
Фенілкетонурія - це спадкове захворювання, яке виражається в зниженому розпаді амінокислоти фенілаланіну. Підступна хвороба полягає в тому, що якщо вона присутня, вона існує з народження і, таким чином, призводить до накопичення (накопичення) амінокислота веде.
Приблизно на третьому місяці життя він присутній в таких високих концентраціях, що надає токсичну дію на психічний розвиток дитини. Захворювання засноване на дефекті ферменту, який перешкоджає конверсії, а отже, і руйнуванню фенілаланіну. Щоб вивести амінокислоту все одно, вона поєднується з різними іншими речовинами в організмі. Виведення цих сполук призводить до особливого неприємного запаху.

Причини фенілкетонурії
Фенілкетонурія викликається двома дефектами ферментів. Ген із "схемою" для ферментів розташований на дванадцятій хромосомі. На сьогоднішній день відомо кілька сотень мутацій цього гена, які викликають захворювання.
Залежно від ступеня мутації, невеликий відсоток ферментів може правильно функціонувати. Залежно від того, наскільки виражений дефіцит ферменту, це призводить до зменшення або неіснуючого розпаду незамінної амінокислоти фенілаланіну. Порівняно з нормальними значеннями концентрації в крові можуть бути в 10 - 20 разів вищими. Частота варіюється в різних етнічних групах і її можна знайти приблизно у кожної десятитисячної дитини в Німеччині.
Накопичення фенілаланіну потім призводить до непоправної шкоди нервовій системі дітей або немовлят.Якщо хворобу не помітити вчасно, у дитини буде спостерігатися розумова відсталість (затримка психологічного та фізичного розвитку), яка погіршиться, поки рівень фенілаланіну не знизиться до нормального рівня.
Детальніше по темі: Етапи раннього розвитку дитини
Як успадковується фенілкетонурія?
Успадкування клінічної картини фенілкетонурії (ПКУ) слідує за аутосомно-рецесивною схемою успадкування, тобто. обидві хромосоми хромосомної пари повинні бути уражені, щоб захворювання стало симптоматичним. Вражений ген, так званий ген PAH, розташований на дванадцятій хромосомі. Зараз відомо кілька сотень типів мутацій, які можуть викликати різні форми фенілкетонурії.
Форми відрізняються залишковою активністю ферменту. Це означає, що певні залишкові кількості фенілаланіну ще можуть бути перетворені. Більшість постраждалих є там гетерозиготний (змішаний). Простіше кажучи, це означає, що не всі клітини несуть хворий ген. Лише невелика частка постраждалих знає генетичний дефект у всіх своїх хромосомах. Існують також регіональні відмінності у типах мутації. У різних регіонах / районах різні варіанти мутацій трапляються по-різному.
Детальніше по темі: Генетичні захворювання
Наскільки часто зустрічається фенілкетонурія?
У глобальному масштабі також існують визначні відмінності щодо захворюваності на захворювання серед населення.
У той час як у Скандинавії та азіатських країнах приблизно фенілкетонурія (ПКУ) страждає приблизно кожні 100 000 людей, країни Балкан, наприклад, демонструють частоту до 1: 5000.
У німецькомовних країнах частота фенілкетонурії становить близько 1: 10 000.
Діагностика фенілкетонурії
Стандартний діагноз встановлюється двома різними способами, один - виявлення дефектного ферменту, а другий - виявлення сильно підвищеної концентрації фенілаланіну в крові.
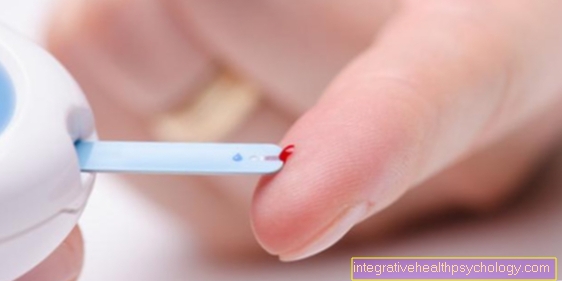
Перший метод, відомий як тандемна мас-спектроскопія, є частиною обстеження новонароджених і показує дефект без пенілаланіну, який не потребує споживання. За допомогою тандемної мас-спектроскопії можна встановити досить надійно понад двадцять інших захворювань.
Другий метод - це тест Гутрі (також його називають тестом на п’яті). Однак обов’язковою умовою є те, що дитина, мабуть, вже вжила фенілаланін - наприклад, через грудне молоко. Під час тесту трохи крові відбирають з п'яти дитини як частину U2 і досліджують на вміст її фенілаланіну.
Детальніше по темі: Огляд U2
За цими симптомами можна розпізнати фенілкетонурію
Симптоми фенілкетонурії значною мірою залежать від кількості залишкової активності ферменту. Якщо ще є певний відсоток функціонуючих ферментів, захворювання не таке виражене, як при вираженій повномасштабній фенілкетонурії.
Типовими симптомами, які помітні на ранній стадії, є сильний та своєрідний запах сечі уражених дітей та психічне та психомоторне недорозвинення дітей у міру її прогресування. Якщо хворобу не виявити і не лікувати рано, відбувається постійне ураження розвитку мозку. У гіршому випадку, діти з їх коефіцієнтом інтелекту можуть залишатися нижче 50 балів IQ.
Крім того, фенілкетонурія може призвести до рухового дефіциту. Важлива сигнальна речовина дофамін відсутня, як це стосується хвороби Паркінсона. Ось чому в цьому контексті можна говорити про так званих неповнолітніх (тобто підлітків) Паркінсона.
Детальніше по темі: Коли дитина повзає? - ви повинні це знати!
Крім того, діти з фенілкетонурією зазвичай мають світле волосся і блакитні очі, оскільки їх виробництво пігменту меланіну також порушено.
Детальніше по темі: Як розпізнати проблеми поведінки у немовлят
Лікування фенілкетонурії
Хороша новина щодо терапії фенілкетонурією полягає в тому, що симптоми психічного недорозвинення можна повністю запобігти, якщо терапію розпочати рано. Основоположним складовою терапії є дієта з низьким вмістом фенілаланіну, яка запобігає накопиченню занадто великої кількості фенілаланіну. Чим пізніше поставлений діагноз і чим пізніше розпочато терапію, тим більша спричинена розумова відсталість (недорозвинення).
Якщо хвороба - це не повна картина фенілкетонурії, а форма зі зниженою активністю ферментів, також може бути проведена так звана замісна терапія. Постраждалі постачаються молекулою тетрагідробіоптерину. Це підтримує організм у перетворенні фенілаланіну. Мірилом терапевтичного успіху є концентрація фенілаланіну в крові. Його слід утримувати в межах від 2 до 4 мг / дл у людей з фенілкетонурією.
Вас також може зацікавити ця тема: Дієта дитини - Рекомендація для немовлят
Що таке дієта з низьким вмістом фенілаланіну?
Як вже було сказано, дієта з низьким вмістом фенілаланіну є наріжним каменем лікування фенілкетонурії. Оскільки фенілаланін є важливою амінокислотою, його не можна повністю уникнути. Це означає, що немовлятам та малюкам потрібно спеціальне молоко для немовлят, оскільки нормальне грудне молоко містить занадто багато фенілаланіну.
Рекомендується продовжувати дієту хоча б до чотирнадцяти років, щоб забезпечити нормальний розумовий розвиток дитини. Оскільки фенілаланін присутній майже у всіх тваринних білках, для хворих рекомендується дієта з низьким вмістом м'яса та м'яса.
Прогноз проти Тривалість життя при фенілкетонурії
Тривалість життя постраждалих залежить від одного боку від типу фенілкетонурії, а з іншого - від часу діагностування захворювання.
Хоча нормальний вік також можливий при нормальному варіанті фенілкетонурії, є рідкісні варіанти захворювання, які пов'язані, наприклад, зі значно підвищеним потенціалом судом, що може призвести до більш смертельних наслідків.
Детальніше по темі: Судоми у дитини
Однак у звичайному варіанті фенілкетонурії вік, мабуть, незначно впливає. Однак час постановки діагнозу і початок лікування визначають, наскільки важкими є психічні порушення хворої людини. Тому розумовий інтелект має максимально опосередкований вплив на тривалість життя.
Прогресування хвороби фенілкетонурії
Перебіг захворювання також різниться, навіть більше, як і тривалість життя залежно від форми захворювання та часу постановки діагнозу або початку лікування.
Якщо захворювання діагностується в скринінгу для новонароджених або на U2, і дієта з низьким вмістом фенілаланіну суворо дотримується, і будь-яке заміщення, яке може стати необхідним, проводиться, захворювання навряд чи з’явиться.
Крім того, люди, які все ще мають залишкову активність ураженого ферменту, мають перевагу, оскільки захворювання у них легке, навіть у гіршому випадку.
Однак якщо діагноз буде поставлений пізно, слід очікувати незворотних психічних розладів, які триватимуть все життя.
Тим не менш, фенілкетонурія - це не захворювання, яке в західних країнах означає передчасну смерть, а скоріше нормальну тривалість життя.
Чи є ліки?
Повноцінне вилікування від хвороби неможливо, оскільки це спадкове захворювання. Максимальна терапія - це дієта з низьким вмістом фенілаланіну, що призводить до зменшення симптомів.
Якщо хвороба розпізнається рано і лікується, навряд чи виникають відхилення; однак особливий запах сечі та токсичний на клітини вплив надмірно високого рівня фенілаланіну виникають знову і знову, як тільки уникнути дієти. Однак, оскільки захворювання успадковується рецесивним чином, малоймовірно, що хвороба передасться.




.jpg)