гемофілія
Синоніми
Гемофілія, спадкове порушення кровотечі, дефіцит фактора згортання крові,
Дефіцит фактора VIII, дефіцит фактора IX, гемофілія
визначення
Гемофілія - це спадкове захворювання системи згортання крові:
У хворих спостерігається порушення згортання крові, що проявляється в тому, що кровотеча відбувається при найменшій травмі, а кровотечу важко зупинити.
Фактори згортання неможливо активувати, тому регулярний хід каскаду згортання неможливий.
Гемофілія - це вроджене захворювання, яке протікає у двох формах,
Гемофілія А і В, може бути присутнім.
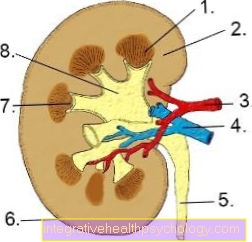
Гемофілія А зустрічається частіше (у 85% випадків), ніж форма В (приблизно 15%) і є більш серйозною з двох форм.
Активність комплексу фактора VIII обмежена при гемофілії А, оскільки у клітинах печінки утворюється фактор VIII-С.
Фактор VIII-C є однією з двох субодиниць комплексу фактора VIII:Віллебранд-Фактор і фактор VIII-С утворюють цей комплекс, що сприяє згортанню крові, в каскаді згортання. Коефіцієнт фон Віллебранда захищає фактор VIII-C від руйнування занадто швидко.
Отже, кількість A. функціонального фактора VIII зменшується у формі А.
В гемофілія Б. синтез фактора згортання IX відбувається в меншій мірі, так що його дія в процесі згортання відсутня, а гемостаз погіршується.
Виникнення / епідеміологія
Виникнення гемофілії у популяції - це гемофілія А. 1 на 5000 і для форми Б. 1:15.000.
Той факт, що чоловіки, зокрема, хворіють, пов’язаний із гендерними проблемами
Х-пов'язана форма успадкування дефекту згортання (див. Причину / розвиток).
Основи
Згортання крові - це тонко налагоджена система: різні компоненти крові, так звані фактори згортання, активуються одна за одною каскадно, щоб уникнути гемостазу у випадку травми.
Коагуляційна система складається з первинного та вторинного гемостазу, який ще називають плазматичною коагуляцією.
Якщо травмується стінка судини, уражена судина звужується на першому кроці (= первинна коагуляція). Тоді опора (=Тромбоцитарний тромб) на травмованій ділянці, яка вкрита тромбоцитами (= Тромбоцити) формується з метою досягнення початкового ущільнення судини.
Утворення тромбу активує плазматичну згортання:
Запускається каскад згортання і взаємодія різних факторів згортання створює стабільну рану або закриття судин.
Причина та походження
The гемофілія Обидві форми гемофілії успадковуються як рецесивна ознака, пов'язана з Х.
У так званому Х-зв'язаному режимі успадкування гемофілії генетична інформація про генний продукт знаходиться на статевій хромосомі Х, яка виявляється у подвійній кількості у жінок та в одиничних числах у чоловіків.
Для кожної генетичної інформації є два гени, так звані алелі. Якщо є спадкове спадкування, обидва алелі повинні бути порушені мутацією, щоб викликати захворювання: рецесивний означає, що патологічно змінена генетична інформація походить від здорової, тобто не змінюється, інформація пригнічується і не призводить до захворювання. Рецесивне спадкове захворювання розвивається лише в тому випадку, якщо обидва алелі несуть неправильну генетичну інформацію.
Є тільки а Вираз гена змінився, давай Ні до гемофілія.
Інформація: спадщина
Якщо змінити лише одну експресію гена, захворювання не буде.
Оскільки у чоловіків є лише одна Х-хромосома в парі статевих хромосом, і тому інформація про генні продукти на їх Х-хромосомі доступна лише у простій формі, вони захворіють, якщо бракує лише один алель.
З іншого боку, жінки, які мають дві Х-хромосоми як статеві хромосоми, хворіють лише тоді, коли обидві хромосоми мають несправні гени для генного продукту.
У чоловіків, які страждають гемофілією, алель, що містить спадкову інформацію про фактор згортання VIII-C (гемофілія А) або фактор згортання IX (гемофілія В), доступний лише у дещо зміненій формі;
Жінки повинні перенести цю генетичну зміну на обох алелях, щоб захворіти.
Якщо у жінок є несправний і не змінений алель, їх називають носіями спадкового захворювання, пов'язаного з Х, наприклад гемофілією.
При гемофілії несправна генетична інформація впливає на зниження активності ураженого фактора згортання або зменшення вироблення фактора згортання.
Хромосома Х є носієм генетичного матеріалу для факторів згортання вторинного гемостазу.
Прибл. 70% мутацій в генетичному матеріалі, які викликають гемофілію, передаються в сім'ї, 30% захворювань виникають внаслідок спонтанних змін (= спонтанні мутації) в генетичній інформації.
діагностика
Після запитання про історію хвороби пацієнта та проведення ретельного фізичного обстеження, є додаткові кроки в діагностиці гемофілії:
У 2/3 випадків у сім’ї трапляються випадки гемофілії, через що слід звернутися до сімейного скупчення захворювання, якщо пацієнт представляє лікаря із симптомами, підозрілими на гемофілію.
Пацієнти повідомляють про синці внаслідок найменших травм.
Один з них відрізняє Тяжкість гемофілії в легку, середню або важку форму захворювання.
Обстеження зразка крові дає такі результати, характерні для гемофілії:
Час кровотечі є нормальним (= первинна згортання недоторканою), але функція плазматичної коагуляції знижується, через що так званий час ПТГ довший.
ПТТ означає частковий час тромбопластину. Він вимірюється в лабораторних тестах для перевірки функції факторів I, II, V, VIII до XII, а також XIV і XV.
Оскільки в гемофілії відсутній один фактор каскаду згортання, він не може працювати оптимально, що призводить до тривалого часу згортання.
Для того, щоб можна було розмежовувати гемофілію А та В, кров пацієнта необхідно досліджувати на фактори VIII та IX, відсутність двох факторів визначає тип гемофілії.
Диференціальний діагноз
Гемофілію потрібно відрізняти від інших порушень системи згортання, зокрема, зокрема, згадується синдром фон Віллебранда-Юргена.
Коефіцієнт фон Віллебранда (vWF) працює разом із фактором VIII-C im
Комплекс фактора VIII і викликає пригнічення передчасної деградації фактора VIII, крім того, фактор сприяє згортанню, опосередковуючи адгезію тромбоцитів крові до травмованого судинного ділянки. Таким чином, vWF є важливою частиною як первинної, так і вторинної коагуляції.
Якщо присутній синдром фон Віллебранда-Юргена, то формування
vWF в клітинах стінки судин відбуваються лише в незначній мірі або неправильним чином. Знижений рівень освіти або неадекватна функція
Фактор фон Віллебранда обумовлений мутаціями в гені для формування фактора. Успадкування синдрому або мутації збудника гена успадковується автосомно-домінантно: генетична інформація для vWF знаходиться на хромосомі №12, яка не є статевою хромосомою. У домінантному режимі успадкування хворий алель пригнічує дію другого здорового алеля, так що захворювання викликається мутацією гена і викликає клінічні симптоми.
Зазвичай пацієнти страждають від шкірних і слизових кровотеч, рідше від спонтанних кровотеч, як у пацієнтів з гемофілією.
Захворювання часто помічають лише при тривалій кровотечі після операції.
Час кровотечі пацієнта після травми тривалий і значення плазматичної згортання патологічно змінені (тривала ПТТ).
Синдром фон Віллебранда-Юргенса викликається прийомом десмопресину або заміною фактора VIII і vWF для стабілізації згортання (більш детальне пояснення див. У розділі «Терапія гемофілії»).
Які симптоми гемофілії?
Симптоми у формах гемофілії не відрізняються:
- Гемофілія зазвичай призводить лише до клінічних симптомів у чоловіків.
- Надмірна кровотеча виникає при гемофілії, що не стосується попереднього, переважно банального нещасного випадку (травма) час кровотечі в нормі, але зазвичай виникає кровотеча, яке не виникає у здорових людей.
- Можна виділити три форми гемофілії, які визначаються залишковою активністю уражених факторів згортання:
1.важка гемофілія (гемофілія) із залишковою активністю менше 1% або часткою функціонального фактору, який все ще присутній, при якому відбувається спонтанна кровотеча та виникає суглобна кровотеча
2. Помірна гемофілія (захворювання крові) з факторною активністю в діапазоні між
1 і 5% нормальної факторної активності та виникнення синців (= гематоми) після незначної травми
3. легка гемофілія (гемофілія), в якій залишається ще 5-15% залишкової активності і при якому пацієнти повідомляють про такі симптоми, як гематоми (синці) після значної травми та післяопераційних кровотеч.
Детальніше по темі: Які причини церебрального крововиливу
- Жінки, які зазвичай мають лише змінену генетичну інформацію щодо гемофілії, зазвичай не виявляють клінічних симптомів із залишковою активністю понад 50%.
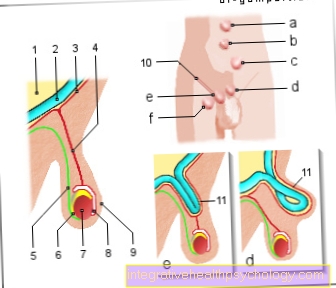
- У пацієнтів спостерігається кровотеча у великі суглоби коліна-, Стегна- або Плечовий суглоб, внаслідок чого можна говорити про гемартроз. Кровотеча запускає запальну реакцію з відновними процесами в суглобі, що може призвести до жорсткості суглоба.
- Він також може кровоточити в Мускулатура і приходять м’які тканини. Кровотеча призводить до підвищення тиску в м’язах або тканині, оскільки зараз є більше обсягу. Особливо це може вплинути на ваші руки і ноги Куперний синдром вести:
Підвищений тиск викликає здавлення судин і нервів, так що кінцівки недостатні, і великі ділянки тканини можуть загинути. Відсічний синдром потрібно якомога швидше лікувати хірурга, щоб запобігти втраті кінцівки. - Виникає кровотеча в живіт, що є небезпечною для життя ситуацією для пацієнта.
- Незвичайно тривала кровотеча можлива після операції.
Крім того, можуть бути тривалі періоди з кров’ю в сечі. Тут один загрожує Анеміятому що пацієнт постійно втрачає кров, можливо, непомітно. - Особливо небезпечні Церебральний параліч (= внутрішньочерепна кровотеча), що призводить до смерті у 10% людей з гемофілією. Ви можете дізнатися більше про цю тему в нашій темі Церебральний параліч.
Як проводиться терапія гемофілії?

Кровотеча має виникати у хворих гемофілія необхідно уникати, через що пацієнт цього не робить Ліки які інгібують згортання крові, такі як Ацетилсаліцилова кислота (Аспірин®) і немає внутрішньом’язових ін'єкцій (= шприци в Мускулатура) відповідно.
Якщо трапляється травма з кровотечею, велике значення має ретельний локальний гемостаз для запобігання кровотечі в сусідні тканини.
Медикаментозна терапія гемофілія полягає в заміні факторів згортання, які організм не може виробляти у достатній кількості.
Хворі з незначним гемофілія отримують препарати фактора згортання у міру необхідності, тобто якщо відбулася травма з кровотечею або планується велика операція, що може призвести до рясної кровотечі.
Профілактичну заміну фактора слід застосовувати пацієнтам із помірною та вираженою гемофілією, тобто відсутній фактор повинен бути наданий постійно, раніше виникає кровотеча.
Якщо залишкова активність фактора VIII перевищує 15% або фактор IX більше 20-25%, регулярна терапія не потрібна; ці пацієнти отримують відсутній фактор згортання у разі спонтанної кровотечі або перед плановою операцією.
Довготривала терапія, а також те, що до операції, може набувати форми домашнього самолікування, при якому пацієнт сам застосовує відсутній фактор згортання.
Для пацієнтів із легкою гемофілією існує альтернатива терапії:
Діюча речовина Десмопресин (наприклад, Minirin®) призводить до розподілу
Фактор VIII, який зберігається в стінках посудини.
Однак препарат можна давати лише протягом декількох днів за раз, оскільки після стимуляції вивільнення фактора пам’ять у стінках судин вичерпується і доводиться повторювати.
У гострій терапії гемофілія Є три варіанти втручання:
- Пацієнт отримує активований протромбін, речовина, що сприяє згортанню крові, щоб кров зупинилася якомога швидше.
- Ще один терапевтичний варіант - прийом препаратів фактора VII. Цей фактор знаходиться на початку каскаду згортання і ініціює його регулярний хід.
- Третій терапевтичний варіант - застосування тваринного фактора VIII-С, щоб дати можливість пацієнту зупинити кровотечу.
Ускладнення
Замінник подарунка (=підміна) факторів згортання може призвести до утворення антитіл проти саме цих факторів, так що заміщення в постійній дозі більше не має терапевтичних ефектів, через що можна говорити про утворення інгібіторів.
Залежно від визначення концентрації антитіл у крові пацієнта, можна вводити високі дози фактора VIII, метою яких є відновлення толерантності до цього фактору та відключення утворення антитіл. Цю процедуру слід проводити лише в спеціалізованих центрах.
Існує низький ризик захворіти гепатитом або заразитися вірусом ВІЛ (=ВІЛ) як фактори походять від продуктів крові людини.
Ще одним ризиком замісної терапії гемофілії є виникнення алергічних реакцій.